Analysis of Ultrashort-Chain and Short-Chain (C1 to C4) PFAS in Water Samples

Abstract
Due to their ubiquitous occurrence in aquatic environments, measuring ultrashort-chain per- and polyfluoroalkyl substances (PFAS) in various source waters to monitor their presence and the potential for human exposure has become very important. However, with carbon chain lengths of less than C4, these small, highly polar compounds are difficult to analyze using standard PFAS tests that are based on reversed-phased liquid chromatography (RPLC). In this study, an accurate, reliable analytical LC-MS/MS method for PFAS in water was developed to specifically quantify C1 to C4 PFAS in both potable and non-potable sources. A direct injection workflow was implemented to simplify the testing process and to avoid potential contamination originating from poor sample preparation procedures.
Introduction
Short-chain PFAS, usually referred to as C4 to C6 compounds, are considered to be less bioaccumulative and thus less toxic than long-chain PFAS. But the practice of using short-chain PFAS as replacements for long-chain PFAS has resulted in their increased environmental accumulation. While the use of short-chain PFAS is intentional, additional studies have shown greater prevalence and higher levels of ultrashort-chain PFAS in environmental aquatic systems (e.g., rain, river, snow, groundwater, and wastewater). The ultrashort-chain PFAS are C1, C2, and C3 compounds, including trifluoroacetic acid (TFA); perfluoropropanoic acid (PFPrA); trifluoromethanesulfonic acid (TFMS); perfluoroethanesulfonic acid (PFEtS); and perfluoropropanesulfonic acid (PFPrS).
Other than TFA, the potential sources and the environmental fate of ultrashort-chain PFAS have not been well studied, partly because limited analytical methods are available to properly monitor their occurrence. GC-MS can be used for the analysis of TFA, but it requires derivatization and does not allow the simultaneous analysis of C1 to C4 perfluoroalkyl carboxylic acids (PFCAs) and perfluoroalkyl sulfonic acids (PFSAs). Anion-exchange LC columns can be used for C1 to C4 PFAS analysis, but due to very strong retention, they produce long runtimes (>20 minutes) and broad peak shapes. Supercritical fluid chromatography (SFC) works well, but the instrument investment can be significant. Reversed-phased liquid chromatography (RPLC) is the predominant analytical approach used to measure PFAS with carbon chain lengths of C4 and up, but RPLC does not have sufficient retention for shorter PFAS. For example, when using RPLC, C4 perfluorobutanoic acid (PFBA) is the least retained short-chain compound, and it often elutes as a broad, asymmetrical peak. Poor peak shape leads to decreased detection sensitivity and less accurate quantification.
To overcome the limitations of RPLC, we previously developed an analytical method for the quantification of C2 to C8 PFAS in both potable and non-potable waters [1]. In that study, we determined that a Raptor Polar X column, with its hybrid HILIC/ion-exchange functionality, was well suited for the simultaneous analysis of legacy, alternative, and ultrashort-chain PFAS. The chromatographic performance of the Raptor Polar X column for C2 to C8 PFAS was characterized by PFSAs eluting earlier than PFCAs. In addition, the compounds with shorter chain lengths were more strongly retained than those with longer chain lengths. In that method, the aqueous mobile phase was 10 mM ammonium formate and 0.05% formic acid in water, and the organic mobile phase was 0.05% formic acid in 60:40 acetonitrile:methanol. The addition of methanol in the organic mobile phase was needed to increase the retention of long-chain PFAS. Furthermore, by using different ratios of acetonitrile and methanol, analyte retention and separation could be controlled in order to mitigate matrix interference. But the use of methanol in the organic mobile phase also had the negative effect of reducing MS sensitivity. In the current study, the original LC-MS/MS method for PFAS in water was reexamined with a focus on only the C1 to C4 PFAS with the goal of increasing detection sensitivity while still maintaining robust performance. Method suitability was evaluated in terms of linearity, accuracy, and precision.
Experimental
Standard and Sample Preparation
The calibration standard solutions (0.4 mL) were prepared in reverse-osmosis (RO) water across a range of 2.5 to 800 ng/L (ppt) in polypropylene HPLC vials. Isotopically-labeled PFBA and perfluorobutanesulfonic acid (PFBS) were used as the internal standards (IS), and an aliquot of 4 µL from a 10 ng/mL working solution was mixed into the standard solutions. Tap water and bottled water were used directly without filtration. Wastewater samples (~10 mL) were filtered with Norm-Ject syringes (10 mL Luer Lock tip; cat.# 22775) and nylon syringe filters (30 mm, 0.45 µm; cat.# 23981) and collected in 50 mL polypropylene tubes. For accuracy and precision experiments, 1 mL water samples were fortified at 25, 50, and 175 ppt, and then a 0.4 mL aliquot was transferred to a polypropylene vial and mixed with 4 µL of the IS working solution before injecting for LC-MS/MS analysis.
LC-MS/MS Method for PFAS in Water
Analysis of PFAS in water samples was performed on a Waters ACQUITY I-class UPLC system coupled to a Xevo TQ-S triple quadrupole mass spectrometer. Compound tuning was conducted using negative mode electrospray ionization to determine the precursor and product ions. The product ion with the strongest intensity was used as the quantifier for concentration determination. A secondary product ion (qualifier) was used for confirmation of the targeted PFAS. There was no qualifier for TFA, PFPrA, and PFBA because only one product ion could be identified. The retention times and MS transition parameters for each analyte are provided in Table I.
Note that while a PFAS delay column is necessary to reduce background contamination of long-chain PFAS from the LC system in gradient elution RPLC methods, it is not needed for this isocratic analysis of the C1 to C4 compounds on a HILIC/ion-exchange Raptor Polar X column.
|
Column:
|
Raptor Polar X (2.7 µm, 50 mm x 2.1 mm ID [cat.# 9311A52])
|
||||||
|
Column temp.:
|
40 °C
|
||||||
|
Injection volume:
|
10 µL
|
||||||
|
Mobile phase A:
|
Water, 10 mM ammonium formate, 0.1% formic acid
|
||||||
|
Mobile phase B:
|
Acetonitrile:isopropanol (95:5), 0.1% formic acid
|
||||||
|
|||||||
|
Flow rate:
|
0.3 mL/min
|
||||||
|
Ion mode:
|
Negative ESI
|
||||||
|
Mode:
|
MRM
|
Table I: MS Ion Transitions, Parameters, and Chromatographic Retention Time of Analytes
| Compounds | Retention Time (min) | Precursor Ion | Product Ions* | Cone (V) | Collision (V) |
| PFBS | 1.01 | 298.97 [M-H]- | 79.97/98.89 | 2 | 26/26 |
| PFPrS | 1.06 | 248.97 [M-H]- | 79.91/98.91 | 2 | 24/24 |
| PFEtS | 1.12 | 198.90 [M-H]- | 79.92/98.91 | 38 | 22/22 |
| TFMS | 1.25 | 148.97 [M-H]- | 79.93/98.92 | 62 | 18/18 |
| PFBA | 1.93 | 213.03 [M-H]- | 168.98 | 14 | 8 |
| PFPrA | 2.23 | 162.97 [M-H]- | 119.02 | 22 | 10 |
| TFA | 3.05 | 113.03 [M-H]- | 69.01 | 10 | 10 |
| 13C3-PFBS | 1.01 | 301.97 [M-H]- | 79.97 | 2 | 28 |
| 13C4-PFBA | 1.93 | 217.03 [M-H]- | 171.98 | 2 | 8 |
* Quantifier ion/qualifier ion
Results and Discussion
Cleanliness of Solvents and Materials for Ultrashort-Chain PFAS Contamination
As ultrashort-chain PFAS are ubiquitous, it is particularly critical to examine the cleanliness of laboratory reagents and materials to ensure they are not contributing background contamination of ultrashort-chain PFAS. Upon testing several brands of LC/MS or HPLC grade water, acetonitrile, isopropanol, and methanol, it was shown that all these solvents had trace or higher amounts of TFA. There were variable amounts of detectable PFPrA and PFBA in the solvents as well. In the end, the commercial solvents with the lowest detectable levels of TFA, PFPrA, and PFBA were selected for the preparation of mobile phases in this study. Deionized water and reverse-osmosis (RO) purified water (generated at Restek) were also tested for cleanliness with regard to the target analytes. The results indicated that the RO water was free of PFPrA and PFBA and contained barely detectable levels of TFA. Therefore, the RO water was used for the preparation of mobile phases, working and final calibration standards, and sample dilutions.
Chromatographic Method Development
When testing a 100% acetonitrile organic mobile phase during method development, the detection signal for C1 to C4 PFAS increased significantly, but this was accompanied by low retention of all analytes. Proper retention was achieved by adjusting the flow rate and the formic acid concentration in the mobile phases. In addition, mixing a small portion of isopropanol with acetonitrile resulted in a stable chromatographic baseline for TFA. For the final method, an aqueous mobile phase of 10 mM ammonium formate plus 0.1% formic acid in RO water (mobile phase A) and an organic mobile phase of 0.1% formic acid in 95:5 acetonitrile:isopropanol (mobile phase B) were used with an isocratic elution under 85% B and flow rate of 0.3 mL/min.
Two isotopically labeled C4 PFAS standards, 13C3-PFBS (M3PFBS) and 13C4-PFBA (M4PFBA), were examined to determine if their use as internal standards was appropriate to compensate for matrix effects. The results showed that the accuracy of quantification was greatly improved by implementing M4PFBA and M3PFBS as the internal standard for the PFCA and PFSA compound classes, respectively. A 5-minute cycle time was appropriate for the standard solution prepared in RO water. However, a 7-minute cycle time was necessary for the analysis of tap water, bottled water, and wastewater to avoid matrix suppression upon repeat injections. Figure 1 shows a chromatogram obtained with the established LC method.
Figure 1: Chromatogram of a C1-C4 PFAS standard (400 ng/L) analyzed under the final LC-MS/MS method conditions.
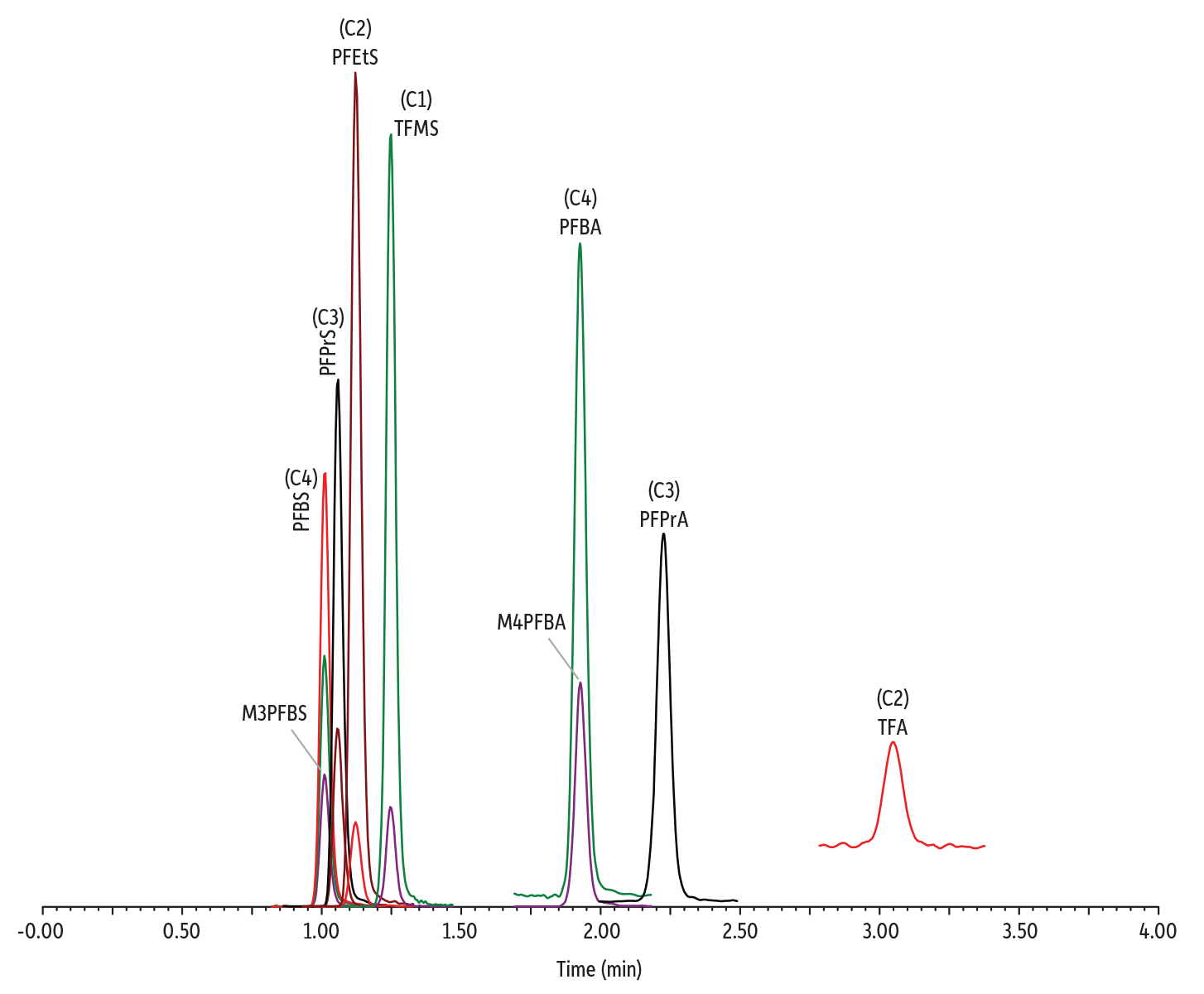
| Column | Raptor Polar X (cat.# 9311A52) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dimensions: | 50 mm x 2.1 mm ID | ||||||||||||
| Particle Size: | 2.7 µm | ||||||||||||
| Pore Size: | 90 Å | ||||||||||||
| Temp.: | 40 °C | ||||||||||||
| Standard/Sample | |||||||||||||
| Diluent: | Reverse osmosis water | ||||||||||||
| Conc.: | 100 - 400 ng/L (ppt) | ||||||||||||
| Inj. Vol.: | 10 µL | ||||||||||||
| Mobile Phase | |||||||||||||
| A: | Water, 10 mM ammonium formate, 0.1% formic acid | ||||||||||||
| B: | 95:5 Acetonitrile:isopropanol, 0.1% formic acid | ||||||||||||
|
| Detector | Waters Xevo TQ-S |
|---|---|
| Ion Mode: | ESI- |
| Mode: | MRM |
| Instrument | Waters ACQUITY UPLC I-Class |
| Sample Preparation | The standard solution was prepared in reverse osmosis water at 400 ppt. An aliquot of 0.4 mL was transferred to a polypropylene HPLC vial and mixed with 4 µL of internal standard solution (10 ng/mL) containing M3PFBS and M4PFBA. |
Linearity
With linear regression (1/x weighted), all analytes showed acceptable linearities with r2 values >0.995 and deviations of <20% across calibration ranges of 2.5–800 ppt for the C1 to C4 PFSA compounds; 5.0–800 ppt for PFBA and PFPrA; and 20–800 ppt for TFA.
Method Accuracy and Precision
Tap water, bottled spring water, and publicly owned treatment works (POTW) wastewater (sewage treatment plant effluent) samples were fortified at 25, 50, and 175 ppt for all analytes. Three batches were analyzed on three different days giving a total of nine replicates at each fortification level for each water sample.
Different incurred amounts of TFA were present in all three water types. The tap water sample contained TFMS as well. In addition to TFA and TFMS, the POTW water sample contained PFBS and PFPrA. These incurred concentrations were subtracted from the calculated concentrations of fortified samples to determine the recovery. Due to a much higher TFA concentration in the POTW water sample, it was diluted fivefold in RO water prior to fortification for determining TFA accuracy and precision. Data were not collected for TFA from 25 ppt fortified samples as accurate quantification by concentration subtraction could not be obtained due to the high levels of incurred TFA. The average recovery and relative standard deviation (RSD) data are shown in Table II. All analytes had recovery values of 86.6–107% across three fortification levels and three different types of waters. Satisfactory method precision was demonstrated by RSD values being within 1.62–10.7%.
Measurement of C1 to C4 PFAS in Potable and Non-Potable Waters
Following the method verification experiments, this direct injection workflow was applied to the determination of C1 to C4 PFAS in a variety of source waters, including tap waters, bottled waters, natural spring water, well water, and wastewaters. Three preparations of blank and fortified (50 ppt) samples were injected for analysis. It was shown that the average recoveries of fortified QC samples were all within 75 to 120%. This demonstrated that the established LC-MS/MS method for PFAS in water was suitable for accurate measurement of C1 to C4 PFAS in both potable and non-potable sources.
Test results for incurred levels are shown in Table III. The data indicated that TFA was ubiquitously present in tap waters at a range of ~120 to 500 ppt. TFMS was present and quantifiable in most tap waters, and PFPrA was detectable in several tap water samples. The bottled spring water samples contained TFA as well. RO purified bottled water and RO purified tap water did not contain detectable levels of C1 to C4 PFAS. The well water sample had a relatively high amount of TFMS compared to the other samples. The wastewater samples, which originated from a POTW facility, a hospital, a metal finisher, and a chemical manufacturer, all had higher levels of TFA and PFPrA. The wastewater effluent collected from the chemical manufacturer was notably elevated for TFA, PFPrA, and PFBA contamination.
Conclusion
An LC-MS/MS method for PFAS in water was established in this study to provide a unique solution for the determination of ultrashort-chain PFAS and short-chain PFAS in various water matrices. The reported method was demonstrated to be rugged, accurate, and precise. In addition, the use of direct injection and the seven-minute chromatographic analysis time make it easy and fast to perform, allowing higher sample throughput. Most important, this solution offers an effective tool for monitoring the emergent C1-C4 PFAS contaminants in environmental water systems, which can assist scientists in generating guidelines for future regulatory actions.
References
- S.H. Liang, A novel approach for ultrashort-chain PFAS analysis in water samples: direct, simultaneous determination of ultrashort-chain, alternative, and legacy PFAS, Application note EVAN3220A-UNV, Restek Corporation, 2021. articles/a-novel-approach-for-ultrashort-chain-pfas-analysis-in-water-samples
Table II: Method accuracy and precision of fortified water samples.
| Tap Water | ||||||
| 25 ng/L | 50 ng/L | 175 ng/L | ||||
| Analyte | %Recovery | %RSD | %Recovery | %RSD | %Recovery | %RSD |
| TFA | - | - | 98.2 | 7.63 | 97.4 | 6.68 |
| PFPrA | 106 | 3.49 | 107 | 2.26 | 103 | 2.19 |
| PFBA | 99.5 | 4.61 | 100 | 5.09 | 101 | 1.72 |
| TFMS | 87.5 | 1.62 | 95.8 | 5.66 | 96.4 | 3.02 |
| PFEtS | 96.2 | 5.68 | 100 | 7.62 | 96.9 | 3.93 |
| PFPrS | 94.2 | 4.80 | 99.8 | 5.38 | 97.3 | 3.60 |
| PFBS | 98.7 | 4.02 | 102 | 4.92 | 101 | 3.79 |
| Bottled Spring Water | ||||||
| 25 ng/L | 50 ng/L | 175 ng/L | ||||
| Analyte | %Recovery | %RSD | %Recovery | %RSD | %Recovery | %RSD |
| TFA | - | - | 107 | 5.92 | 97.1 | 4.27 |
| PFPrA | 96.6 | 4.1 | 107 | 4.29 | 102 | 2.19 |
| PFBA | 94.4 | 9.17 | 101 | 5.08 | 99.6 | 3.12 |
| TFMS | 86.6 | 5.99 | 95.5 | 5.74 | 94.6 | 3.99 |
| PFEtS | 92.0 | 6.18 | 101 | 6.24 | 95.1 | 6.77 |
| PFPrS | 92.5 | 7.94 | 99.4 | 6.31 | 96.1 | 4.50 |
| PFBS | 95.5 | 8.10 | 104 | 7.03 | 98.6 | 5.09 |
| POTW Water | ||||||
| 25 ng/L | 50 ng/L | 175 ng/L | ||||
| Analyte | %Recovery | %RSD | %Recovery | %RSD | %Recovery | %RSD |
| TFA | - | - | 96.7 | 10.7 | 106 | 4.02 |
| PFPrA | 102 | 3.08 | 102 | 3.02 | 101 | 1.71 |
| PFBA | 100 | 6.36 | 95.2 | 5.25 | 97.4 | 1.62 |
| TFMS | 92.6 | 7.42 | 94.5 | 7.94 | 93.8 | 5.25 |
| PFEtS | 93.8 | 6.54 | 97.2 | 7.75 | 95.7 | 7.48 |
| PFPrS | 97.6 | 4.47 | 97.6 | 6.52 | 96.8 | 5.78 |
| PFBS | 99.8 | 6.97 | 103 | 5.99 | 100 | 3.58 |
Table III: The quantification of C1 to C4 PFAS in various potable and non-potable waters.
| Water Samples | Average Concentration (ng/L; ppt) | ||||||
| TFA | PFPrA | PFBA | TFMS | PFEtS | PFPrS | PFBS | |
| Potable Waters | |||||||
| Tap water #1 | 230 | nd | nd | 5.58 | nd | nd | nd |
| Tap water #2 | 520 | nd | nd | 6.88 | nd | nd | nd |
| Tap water #3 | 450 | <5.00 | nd | 3.20 | nd | nd | nd |
| Tap water #4 (filtered well water) |
267 | nd | nd | nd | nd | nd | nd |
| Tap water #5 | 297 | <5.00 | nd | 4.68 | nd | nd | nd |
| Tap water #6 | 428 | <5.00 | nd | <2.5 | nd | nd | nd |
| Tap water #7 (RO filtered tap water #6) |
nd | nd | nd | nd | nd | nd | nd |
| Tap water #8 | 400 | <5.00 | nd | nd | nd | nd | nd |
| Tap water #9 | 228 | nd | nd | 5.22 | nd | nd | nd |
| Tap water #10 | 117 | nd | nd | nd | nd | nd | nd |
| Bottled water #1 (RO purified) |
nd | nd | nd | nd | nd | nd | nd |
| Bottled water #2 (spring water) |
102 | nd | nd | nd | nd | nd | nd |
| Bottled water #3 (spring water) |
368 | nd | nd | <2.5 | nd | nd | nd |
| Natural spring water | 527 | <5.00 | nd | <2.5 | nd | nd | nd |
| Well water | 342 | nd | nd | 15.6 | nd | nd | nd |
| Non-Potable Waters | |||||||
| POTW water (treated sewage wastewater, effluent) |
1113 | 36.6 | <5.00 | 8.53 | nd | nd | 4.35 |
| Hospital effluent | 1363 | 24.6 | <5.00 | 4.67 | nd | nd | nd |
| Metal finisher | 741 | 11.4 | <5.00 | 5.16 | nd | nd | 2.77 |
| Chemical manufacturer effluent | 131,200 | 11,084 | 52.0 | 4.02 | nd | nd | nd |
nd = not detected